Joint Regression is a very old, but, nonetheless, useful technique. It is widely known that the yield of a genotype in different environments depends on environmental covariates, such as the amount of rainfall in some critical periods of time. Apart from rain, also temperature, wind, solar radiation, air humidity and soil characteristics may concur to characterise a certain environment as good or bad and, ultimately, to determine yield potential.
Early in the 60s, several authors proposed that the yield of genotypes is expressed as a function of an environmental index \(e_j\), measuring the yield potential of each environment \(j\) (Finlay and Wilkinson, 1963; Eberhart and Russel, 1966; Perkins and Jinks, 1968). For example, for a genotype \(i\), we could write:
\[y_{ij} = \mu_i + \beta_i e_j\]
where the yield \(y_i\) in a certain environment \(j\) is expressed as a linear function of the environmental index \(e_j\); \(\mu_i\) is the intercept and \(\beta_i\) is the slope, which expresses how the genotype \(i\) responds to the environment.
A graphical example may be useful; in the figure below we have two genotypes tested in 10 environments. The yield of the first genotype (red) increases as the environmental index increases, with slope \(\beta_1 = 0.81\). On the other hand, the yield of the second genotype (blue) does not change much with the environment (\(\beta_2 = -0.08)\). Clearly, a high value of \(\beta\) demonstrates that the genotype is responsive to the environment and makes profit of favorable conditions. Otherwise, a low \(\beta\) value (close to 0) demonstrates that the genotype is not responsive and tends to give more or less the same yield in all environments (static stability; Wood, 1976).
## `geom_smooth()` using formula = 'y ~ x'
## `geom_smooth()` using formula = 'y ~ x'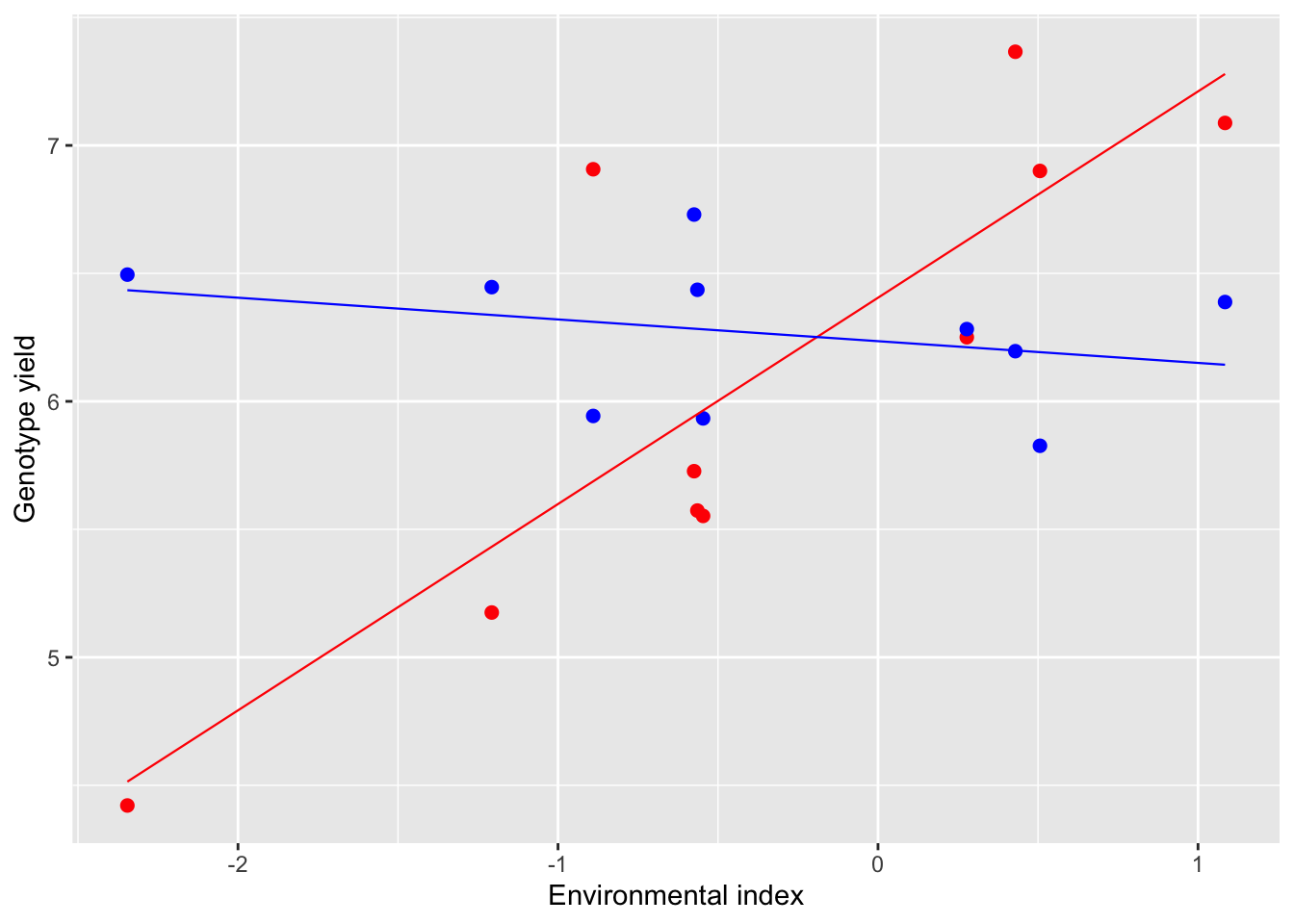
By now, it should be clear that \(\beta\) is a relevant measure of stability. Now, the problem is: how do we determine such value from a multi-environment genotype experiment? As usual, let’s start from a meaningful example.
A multi-environment experiment
Let’s take the data in Sharma (2006; Statistical And Biometrical Techniques In Plant Breeding, New Age International ltd. New Delhi, India). They refer to a multi-environment experiment with 7 genotypes, 6 environments and 3 blocks; let’s load the data in the dataframe ‘dataFull’.
rm(list=ls())
library(nlme)
library(emmeans)
Block <- factor(rep(c(1:3), 42))
Var <- factor(rep(LETTERS[1:7],each=18))
Loc <- factor(rep(rep(letters[1:6], each=3), 7))
P1 <- factor(Loc:Block)
Yield <- c(60,65,60,80,65,75,70,75,70,72,82,90,48,45,50,50,40,40,
80,90,83,70,60,60,85,90,90,70,85,80,40,40,40,38,40,50,
25,28,30,40,35,35,35,30,30,40,35,35,35,25,20,35,30,30,
50,65,50,40,40,40,48,50,52,45,45,50,50,50,45,40,48,40,
52,50,55,55,54,50,40,40,60,48,38,45,38,30,40,35,40,35,
22,25,25,30,28,32,28,25,30,26,28,28,45,50,45,50,50,50,
30,30,25,28,34,35,40,45,35,30,32,35,45,35,38,44,45,40)
dataFull <- data.frame(Block, Var, Loc, Yield)
rm(Block, Var, Loc, P1, Yield)
head(dataFull)
## Block Var Loc Yield
## 1 1 A a 60
## 2 2 A a 65
## 3 3 A a 60
## 4 1 A b 80
## 5 2 A b 65
## 6 3 A b 75What is an environmental index?
First of all, we need to define an environmental index, which can describe the yield potential in each of the seven environments. Yates and Cochran (1937) proposed that we use the mean of all observations in each environment, expressed as the difference between the environmental mean yield \(\mu_j\) and the overall mean \(\mu\) (i.e. \(e_j = \mu_j - \mu\)). Let’s do it; in the box below we use the package ‘dplyr’ to augment the dataset with a new variable, representing the environmental indices.
library(dplyr)
dataFull <- dataFull %>%
group_by(Loc) %>%
mutate(ej = mean(Yield) - mean(dataFull$Yield))
head(dataFull)
## # A tibble: 6 × 5
## # Groups: Loc [2]
## Block Var Loc Yield ej
## <fct> <fct> <fct> <dbl> <dbl>
## 1 1 A a 60 1.45
## 2 2 A a 65 1.45
## 3 3 A a 60 1.45
## 4 1 A b 80 0.786
## 5 2 A b 65 0.786
## 6 3 A b 75 0.786This step is ok with balanced data and it is clear that a high environmental index identifies the favorable environments, while a low (negative) environmental index identifies unfavorable environments. It is necessary to keep in mind that we have unwillingly put a constraint on \(e_j\) values, that have to sum up to zero.
Full model definition (Equation 1)
Now, it is possible to regress the yield data for each genotype against the environmental indices, according to the following joint regression model:
\[y_{ijk} = \gamma_{jk} + \mu_i + \beta_i e_j + d_{ij} + \varepsilon_{ijk} \quad\quad\quad \textrm{(Equation 1)}\]
where: \(y_{ijk}\) is the yield for the genotype \(i\) in the environment \(j\) and block \(k\), \(\gamma\) is the effect of blocks within environments and \(\mu_i\) is the average yield for the genotype \(i\). As we have seen in the figure above, the average yield of a genotype in each environment cannot be exactly described by the regression against the environmental indices (in other words: the observed means do not lie along the regression line). As the consequence, we need the random term \(d_{ij}\) to represent the deviation from the regression line for the genotype \(i\) in the environment \(j\). Finally, the random elements \(\varepsilon_{ijk}\) represent the deviations between the replicates for the genotype \(i\) in the environment \(j\) (within-trial errors). As I said, \(d_{ij}\) and \(\varepsilon_{ijk}\) are random, with variances equal to \(\sigma^2_d\) and \(\sigma^2\), respectively.
According to Finlay-Wilkinson, \(\sigma^2_d\) is assumed to be equal for all genotypes. Otherwise, according to Eberarth-Russel, \(\sigma^2_{d}\) may assume a different value for each genotype (\(\sigma^2_{d(i)}\)) and may become a further measure of stability: if this is small, a genotype does not show relevant variability of yield, apart from that due to the regression against the environmental indices.
Model fitting
We can start the analyses by fitting a traditional ANOVA model, to keep as a reference.
mod.aov <- lm(Yield ~ Loc/Block + Var*Loc, data = dataFull)
anova(mod.aov)
## Analysis of Variance Table
##
## Response: Yield
## Df Sum Sq Mean Sq F value Pr(>F)
## Loc 5 1856.0 371.2 17.9749 1.575e-11 ***
## Var 6 20599.2 3433.2 166.2504 < 2.2e-16 ***
## Loc:Block 12 309.8 25.8 1.2502 0.2673
## Loc:Var 30 12063.6 402.1 19.4724 < 2.2e-16 ***
## Residuals 72 1486.9 20.7
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1As we said, Equation 1 is a mixed model, which calls for the use of the ‘lme()’ function. For better understanding, it is useful to start by augmenting the previous ANOVA model with the regression term (‘Var/ej’). We use the nesting operator, to have different regression lines for each level of ‘Var’.
# Augmented ANOVA model
mod.aov2 <- lm(Yield ~ Loc/Block + Var/ej + Loc:Var, data=dataFull)
anova(mod.aov2)
## Analysis of Variance Table
##
## Response: Yield
## Df Sum Sq Mean Sq F value Pr(>F)
## Loc 5 1856.0 371.2 17.9749 1.575e-11 ***
## Var 6 20599.2 3433.2 166.2504 < 2.2e-16 ***
## Loc:Block 12 309.8 25.8 1.2502 0.2673
## Var:ej 6 9181.2 1530.2 74.0985 < 2.2e-16 ***
## Loc:Var 24 2882.5 120.1 5.8159 2.960e-09 ***
## Residuals 72 1486.9 20.7
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We see that the GE interaction in the ANOVA model has been decomposed into two parts: the regression term (‘Var/ej’) and the deviation from regression (‘Loc:Var’), with 6 and 24 degrees of freedom, respectively. This second term corresponds to \(d_{ij}\) in Equation 1 (please, note that the two terms ‘Var/ej’ and ‘Loc:Var’ are partly confounded).
The above analysis is only useful for teaching purposes, but it is unsatisfactory, because the \(d_{ij}\) terms have been regarded as fixed, which is pretty illogical. Therefore, we change the fixed effect model into a mixed model, where we include the random ‘genotype by environment’ interaction. We also change the fixed block effect into a random effect and remove the intercept, to more strictly adhere to the parameterisation of Equation 1. The two random effects ‘Loc:Block’ and ‘Loc:Var’ are not nested into each other and we need to code them by using ‘pdMat’ constructs, which are not straightforward. You can use the code in the box below as a guidance to fit a Finlay-Wilkinson model.
# Finlay-Wilkinson model
modFull1 <- lme(Yield ~ Var/ej - 1,
random = list(Loc = pdIdent(~ Var - 1),
Loc = pdIdent(~ Block - 1)),
data=dataFull)
summary(modFull1)$tTable
## Value Std.Error DF t-value p-value
## VarA 63.1666667 2.4017164 107 26.3006350 1.624334e-48
## VarB 66.1666667 2.4017164 107 27.5497417 2.135264e-50
## VarC 31.8333333 2.4017164 107 13.2544097 2.599693e-24
## VarD 47.1111111 2.4017164 107 19.6156012 3.170228e-37
## VarE 44.7222222 2.4017164 107 18.6209421 2.378452e-35
## VarF 34.2777778 2.4017164 107 14.2722004 1.614127e-26
## VarG 35.8888889 2.4017164 107 14.9430169 6.028635e-28
## VarA:ej 3.2249875 0.6257787 107 5.1535588 1.176645e-06
## VarB:ej 4.7936139 0.6257787 107 7.6602379 8.827229e-12
## VarC:ej 0.4771074 0.6257787 107 0.7624219 4.474857e-01
## VarD:ej 0.3653064 0.6257787 107 0.5837629 5.606084e-01
## VarE:ej 1.2369950 0.6257787 107 1.9767291 5.064533e-02
## VarF:ej -2.4316943 0.6257787 107 -3.8858692 1.770611e-04
## VarG:ej -0.6663160 0.6257787 107 -1.0647790 2.893729e-01
VarCorr(modFull1)
## Variance StdDev
## Loc = pdIdent(Var - 1)
## VarA 27.5007919 5.2441197
## VarB 27.5007919 5.2441197
## VarC 27.5007919 5.2441197
## VarD 27.5007919 5.2441197
## VarE 27.5007919 5.2441197
## VarF 27.5007919 5.2441197
## VarG 27.5007919 5.2441197
## Loc = pdIdent(Block - 1)
## Block1 0.4478291 0.6692003
## Block2 0.4478291 0.6692003
## Block3 0.4478291 0.6692003
## Residual 20.8781458 4.5692610From the output, we see that the variance component \(\sigma_d\) (27.50) is the same for all genotypes; if we want to let a different value for each genotype (Eberarth-Russel model), we need to change the ‘pdMat’ construct for the ‘Loc:Var’ effect, turning from ‘pdIdent’ to ‘pdDiag’, as shown in the box below.
# Eberhart-Russel model
modFull2 <- lme(Yield ~ Var/ej - 1,
random = list(Loc = pdDiag(~ Var - 1),
Loc = pdIdent(~ Block - 1)),
data=dataFull)
summary(modFull2)$tTable
## Value Std.Error DF t-value p-value
## VarA 63.1666667 3.0507629 107 20.7052032 3.221930e-39
## VarB 66.1666667 2.7818326 107 23.7852798 1.604422e-44
## VarC 31.8333333 1.7240721 107 18.4640387 4.753742e-35
## VarD 47.1111111 2.3526521 107 20.0246824 5.564350e-38
## VarE 44.7222222 2.4054296 107 18.5921974 2.699536e-35
## VarF 34.2777778 1.9814442 107 17.2993906 8.947485e-33
## VarG 35.8888889 2.2617501 107 15.8677515 7.076551e-30
## VarA:ej 3.2249875 0.7948909 107 4.0571447 9.466174e-05
## VarB:ej 4.7936139 0.7248198 107 6.6135249 1.522848e-09
## VarC:ej 0.4771074 0.4492152 107 1.0620909 2.905857e-01
## VarD:ej 0.3653064 0.6129948 107 0.5959372 5.524757e-01
## VarE:ej 1.2369950 0.6267462 107 1.9736777 5.099652e-02
## VarF:ej -2.4316943 0.5162748 107 -4.7100774 7.473942e-06
## VarG:ej -0.6663160 0.5893098 107 -1.1306718 2.607213e-01
VarCorr(modFull2)
## Variance StdDev
## Loc = pdDiag(Var - 1)
## VarA 48.7341240 6.9809830
## VarB 39.3227526 6.2707856
## VarC 10.7257438 3.2750181
## VarD 26.1010286 5.1089166
## VarE 27.6077467 5.2543074
## VarF 16.4479246 4.0556041
## VarG 23.5842788 4.8563648
## Loc = pdIdent(Block - 1)
## Block1 0.4520678 0.6723599
## Block2 0.4520678 0.6723599
## Block3 0.4520678 0.6723599
## Residual 20.8743411 4.5688446From the regression slopes we see that the genotypes A and B are the most responsive to the environment (\(\beta_A = 3.22\) and \(\beta_B = 4.79\), respectively), while the genotypes C and D are stable in a static sense, although their average yield is pretty low.
Fitting a joint regression model in two-steps (Equation 2)
In the previous analyses we used the plot data to fit a joint regression model. In order to reduce the computational burden, it may be useful to split the analyses in two-steps:
- we analyse the plot data, to retrieve the means for the ‘genotype by environment’ combinations;
- we fit the joint regression model to those means.
The results of the two approaches are not necessarily the same, as some information in the first step is lost in the second. Several weighing schemes have been proposed to make two-steps fitting more reliable (Möhring and Piepho, 2009); in this example, I will show an unweighted two-steps analyses, which is simple, but not necessarily the best way to go.
A model for the second step is:
\[y_{ij} = \mu_i + \beta_i e_j + f_{ij} \quad\quad\quad \textrm{(Equation 2)}\]
where the residual random component \(f_{ij}\) is assumed as normally distributed, with mean equal to zero and variance equal to \(\sigma^2_f\). In general, \(\sigma^2_f > \sigma^2_d\), as the residual sum of squares from Model 2 also contains a component for within trial errors. Indeed, for a balanced experiment, it is:
\[\sigma^2_{f} = \sigma^2_d + \frac{\sigma^2}{k}\]
where \(\sigma^2\) is the within-trial error, which needs to be obtained from the first step. In the previous analyses we have already fitted an anova model to the whole dataset (‘mod.aov’). In the box below, we make use of the ‘emmeans’ package to retrieve the least squares means for the seven genotypes in all locations. Subsequently, the environmental means are calculated and centered, by subtracting the overall mean.
library(emmeans)
muGE <- as.data.frame( emmeans(mod.aov, ~Var:Loc) )[,1:3]
names(muGE) <- c("Var", "Loc", "Yield")
muGE <- muGE %>%
group_by(Loc) %>%
mutate(ej = mean(Yield) - mean(muGE$Yield))Now, we fit Equation 2 to the means. In the code below we assume homoscedasticity and, thus, we are fitting the Finlay-Wilkinson model. The variance component \(\sigma^2_d\) is obtained by subtracting a fraction of the residual variance from the first step.
# Finlay-Wilkinson model
modFinlay <- lm(Yield ~ Var/ej - 1, data=muGE)
summary(modFinlay)
##
## Call:
## lm(formula = Yield ~ Var/ej - 1, data = muGE)
##
## Residuals:
## Min 1Q Median 3Q Max
## -7.3981 -3.5314 -0.8864 3.7791 11.2045
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## VarA 63.1667 2.3915 26.413 < 2e-16 ***
## VarB 66.1667 2.3915 27.668 < 2e-16 ***
## VarC 31.8333 2.3915 13.311 1.24e-13 ***
## VarD 47.1111 2.3915 19.699 < 2e-16 ***
## VarE 44.7222 2.3915 18.701 < 2e-16 ***
## VarF 34.2778 2.3915 14.333 2.02e-14 ***
## VarG 35.8889 2.3915 15.007 6.45e-15 ***
## VarA:ej 3.2250 0.6231 5.176 1.72e-05 ***
## VarB:ej 4.7936 0.6231 7.693 2.22e-08 ***
## VarC:ej 0.4771 0.6231 0.766 0.450272
## VarD:ej 0.3653 0.6231 0.586 0.562398
## VarE:ej 1.2370 0.6231 1.985 0.056998 .
## VarF:ej -2.4317 0.6231 -3.902 0.000545 ***
## VarG:ej -0.6663 0.6231 -1.069 0.294052
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 5.858 on 28 degrees of freedom
## Multiple R-squared: 0.9905, Adjusted R-squared: 0.9857
## F-statistic: 208.3 on 14 and 28 DF, p-value: < 2.2e-16
sigmaf <- summary(modFinlay)$sigma^2
sigma2 <- summary(mod.aov)$sigma^2
sigmaf - sigma2/3 #sigma2_d
## [1] 27.43169In the box below, we allow for different variances for each genotype and, therefore, we fit the Eberarth-Russel model. As before, we can retrieve the variance components \(\sigma^2_{d(i)}\) from the fitted model object, by subtracting the within-trial error obtained in the first step.
# Eberarth-Russel model
modEberarth <- gls(Yield ~ Var/ej - 1,
weights=varIdent(form=~1|Var), data=muGE)
coefs <- summary(modEberarth)$tTable
coefs
## Value Std.Error t-value p-value
## VarA 63.1666667 3.0434527 20.7549360 1.531581e-18
## VarB 66.1666667 2.7653537 23.9270177 3.508778e-20
## VarC 31.8333333 1.7165377 18.5450822 2.912238e-17
## VarD 47.1111111 2.3344802 20.1805574 3.204306e-18
## VarE 44.7222222 2.3899219 18.7128381 2.304763e-17
## VarF 34.2777778 1.9783684 17.3262868 1.685683e-16
## VarG 35.8888889 2.2589244 15.8876005 1.537133e-15
## VarA:ej 3.2249875 0.7929862 4.0668898 3.511248e-04
## VarB:ej 4.7936139 0.7205262 6.6529352 3.218756e-07
## VarC:ej 0.4771074 0.4472521 1.0667527 2.951955e-01
## VarD:ej 0.3653064 0.6082600 0.6005761 5.529531e-01
## VarE:ej 1.2369950 0.6227056 1.9864844 5.684599e-02
## VarF:ej -2.4316943 0.5154734 -4.7174004 6.004832e-05
## VarG:ej -0.6663160 0.5885736 -1.1320862 2.672006e-01
sigma <- summary(modEberarth)$sigma
sigma2fi <- (c(1, coef(modEberarth$modelStruct$varStruct, uncons = FALSE)) * sigma)^2
names(sigma2fi)[1] <- "A"
sigma2fi - sigma2/3 #sigma2_di
## A B C D E F G
## 48.69203 38.99949 10.79541 25.81519 27.38676 16.60005 23.73284Fitting in two steps we obtain the very same result as with fitting in one step, but it ain’t necessarily so.
I would like to conclude by saying that a joint regression model, the way I have fitted it here, is simple and intuitively appealing, although it has been criticized for a number of reasons. In particular, it has been noted that the environmental indices \(e_j\) are estimated from the observed data and, therefore, they are not precisely known. On the contrary, linear regression makes the assumption that the levels of explanatory variables are precisely known and not sampled. As the consequence, our estimates of the slopes \(\beta\) may be biased. Furthermore, in our construction we have put some arbitrary constraints on the environmental indices (\(\sum{e_j} = 0\)) and on the regression slopes (\(\sum({\beta_i})/G = 1\); where G is the number of genotypes), which are not necessarily reasonable.
Alternative methods of fitting joint regression models have been proposed (see Piepho, 1998), but they are slightly more complex and I will deal with them in a future post.
Happy coding!
Prof. Andrea Onofri
Department of Agricultural, Food and Environmental Sciences
University of Perugia (Italy)
Borgo XX Giugno 74
I-06121 - PERUGIA
References
- Eberhart, S.A., Russel, W.A., 1966. Stability parameters for comparing verieties. Crop Science 6, 36–40.
- Finlay, K.W., Wilkinson, G.N., 1963. The analysis of adaptation in a plant-breeding programme. Australian Journal of Agricultural Research 14, 742–754.
- Möhring, J., Piepho, H.-P., 2009. Comparison of Weighting in Two-Stage Analysis of Plant Breeding Trials. Crop Science 49, 1977–1988.
- Perkins, J.M., Jinks, J.L., 1968. Environmental gentype-environmental components of variability. III. Multiple lines and crosses. Heredity 23, 339–356.
- Piepho, H.-P., 1998. Methods for comparing the yield stability of cropping systems - A review. Journal of Agronomy and Crop Science 180, 193–213.
- Wood, J., 1976. The use of environmental variables in the interpretation of genotype-environment interaction. Heredity 37, 1–7.
- Yates, F., and Cochran G., 1938. The analysis of groups of experiments. Journal of Agricultural Sciences, 28, 556—580.