ANOVA is routinely used in applied biology for data analyses, although, in some instances, the basic assumptions of normality and homoscedasticity of residuals do not hold. In those instances, most biologists would be inclined to adopt some sort of stabilising transformations (logarithm, square root, arcsin square root…), prior to ANOVA. Yes, there might be more advanced and elegant solutions, but stabilising transformations are suggested in most traditional biometry books, they are very straightforward to apply and they do not require any specific statistical software. I do not think that this traditional technique should be underrated.
However, the use of stabilising transformations has one remarkable drawback, it may hinder the clarity of results. I’d like to give a simple, but relevant example.
An example with counts
Consider the following dataset, that represents the counts of insects on 15 independent leaves, treated with the insecticides A, B and C (5 replicates):
dataset <- structure(data.frame(
Insecticide = structure(c(1L, 1L, 1L, 1L, 1L,
2L, 2L, 2L, 2L, 2L, 3L, 3L, 3L, 3L, 3L),
.Label = c("A", "B", "C"), class = "factor"),
Count = c(448, 906, 484, 477, 634, 211, 276,
415, 587, 298, 50, 90, 73, 44, 26)),
.Names = c("Insecticide", "Count"))
dataset
## Insecticide Count
## 1 A 448
## 2 A 906
## 3 A 484
## 4 A 477
## 5 A 634
## 6 B 211
## 7 B 276
## 8 B 415
## 9 B 587
## 10 B 298
## 11 C 50
## 12 C 90
## 13 C 73
## 14 C 44
## 15 C 26We should not expect that a count variable is normally distributed with equal variances. Indeed, a graph of residuals against expected values shows clear signs of heteroscedasticity.
mod <- lm(Count ~ Insecticide, data=dataset)
plot(mod, which = 1)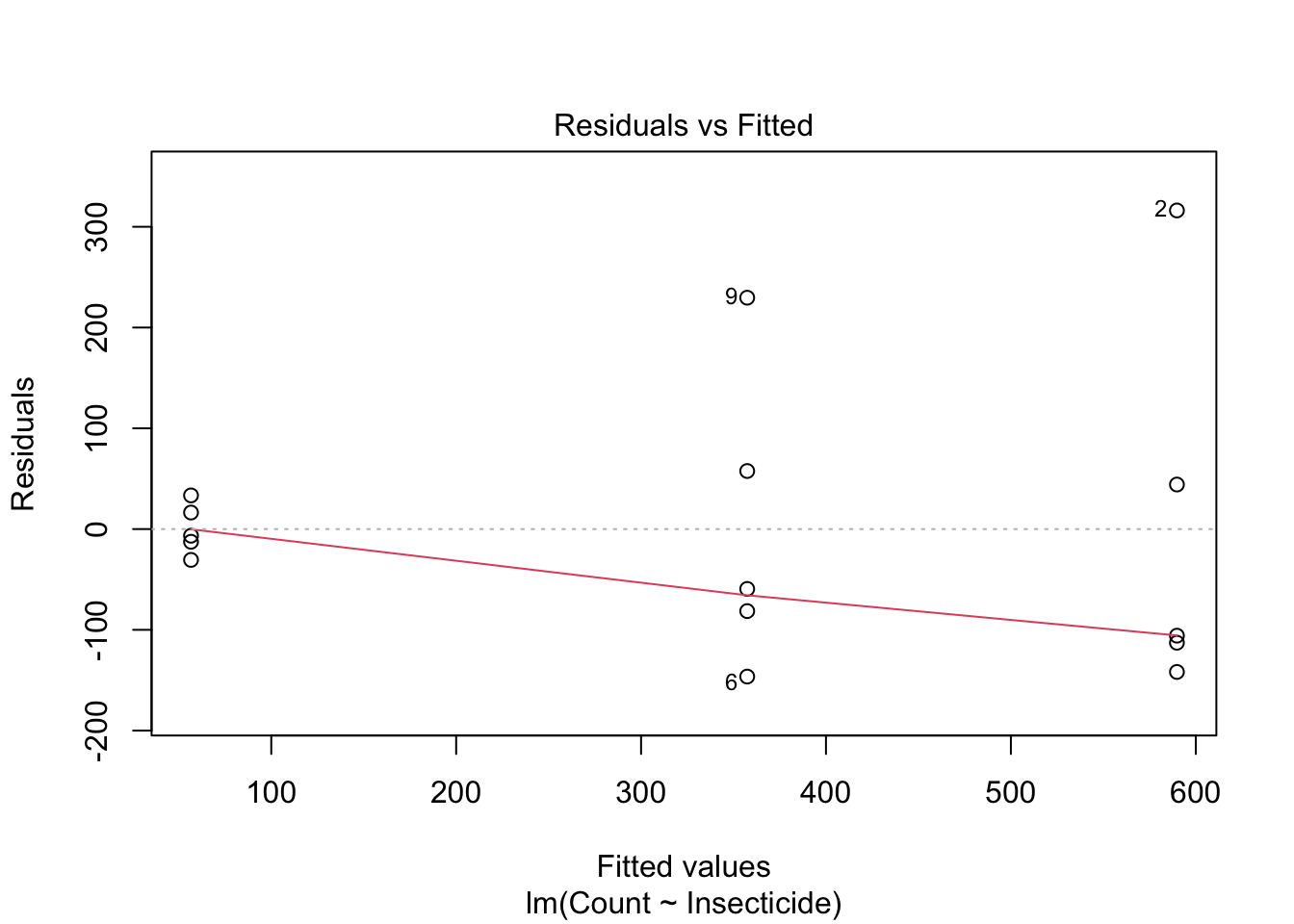
In this situation, a logarithmic transformation is often suggested to produce a new normal and homoscedastic dataset. Therefore we take the log-transformed variable and submit it to ANOVA.
model <- lm(log(Count) ~ Insecticide, data=dataset)
print(anova(model), digits=6)
## Analysis of Variance Table
##
## Response: log(Count)
## Df Sum Sq Mean Sq F value Pr(>F)
## Insecticide 2 15.82001 7.91000 50.1224 1.4931e-06 ***
## Residuals 12 1.89376 0.15781
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
r
summary(model)
##
## Call:
## lm(formula = log(Count) ~ Insecticide, data = dataset)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.6908 -0.1849 -0.1174 0.2777 0.5605
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 6.3431 0.1777 35.704 1.49e-13 ***
## InsecticideB -0.5286 0.2512 -2.104 0.0572 .
## InsecticideC -2.3942 0.2512 -9.529 6.02e-07 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.3973 on 12 degrees of freedom
## Multiple R-squared: 0.8931, Adjusted R-squared: 0.8753
## F-statistic: 50.12 on 2 and 12 DF, p-value: 1.493e-06In this example, the standard error for each mean (SEM) corresponds to \(\sqrt{0.158/5}\). In the end, we might show the following table of means for transformed data:
| Insecticide | Means (log n.) |
|---|---|
| A | 6.343 |
| B | 5.815 |
| C | 3.985 |
| SEM | 0.178 |
Unfortunately, we loose clarity: how many insects did we have on each leaf? If we present in our manuscript a table like this one we might be asked by our readers or by the reviewer to report the means on the original measurement unit. What should we do, then? Here are some suggestions.
- We can present the means of the original data with standard deviations. This is clearly less than optimal, if we want to suggest more than the bare variability of the observed sample. Furthermore, please remember that the means of original data may not be a good measure of central tendency, if the original population is strongly ‘asymmetric’ (skewed)!
- We can show back-transformed means. Accordingly, if we have done, e.g., a logarithmic transformation, we can exponentiate the means of transformed data and report them back to the original measurement unit. Back-transformed means ‘estimate’ the medians of the original populations, which may be regarded as better measures of central tendency for skewed data.
We suggest that the use of the second method. However, this leaves us with the problem of adding a measure of uncertainty to back-transformed means. No worries, we can use the delta method to back-transform standard errors. It is straightforward:
- take the first derivative of the back-transform function [in this case the first derivative of exp(X)=exp(X)] and
- multiply it by the standard error of the transformed data.
This may be simply done by hand, with e.g \(exp(6.343) \times 0.178 = 101.19\) (for insecticide A). This ‘manual’ solution is always available, regardless of the statistical software at hand. With R, we can use the ‘emmeans’ package (Lenth, 2016):
library(emmeans)
countM <- emmeans(model, ~Insecticide, regrid = "response")It is enough to set the argument ‘regrid’ to ’response, although the transformation must be embedded in the model. It means: it is ok if we coded the model as:
log(Count) ~ InsecticideOn the contrary, it fails if we coded the model as:
logCount ~ Insecticidewhere the transformation was performed prior to fitting (there are methods to circumvent this, but I’ll explain in another post).
Obviously, the back-transformed standard error is different for each mean (there is no homogeneity of variances on the original scale, but we knew this already). Back-transformed data might be presented as follows:
| Insecticide | Mean | SE |
|---|---|---|
| A | 568.5 | 101.19 |
| B | 335.1 | 59.68 |
| C | 51.88 | 9.57 |
It would be appropriate to state it clearly (e.g. in a footnote), that means and SEs were obtained by back-transformation via the delta method. Far clearer, isn’t it? As I said, there are other solutions, such as fitting a GLM, but stabilising transformations are simple and they are easily acceptable in biological Journals.
If you want to know something more about the delta-method you might start from my post here. A few years ago, some collegues and I have also discussed these issues in a journal paper (Onofri et al., 2010).
Thanks for reading!
Andrea Onofri
University of Perugia (Italy)
#References
- Lenth, R.V., 2016. Least-Squares Means: The R Package lsmeans. Journal of Statistical Software 69. https://doi.org/10.18637/jss.v069.i01
- Onofri, A., Carbonell, E.A., Piepho, H.-P., Mortimer, A.M., Cousens, R.D., 2010. Current statistical issues in Weed Research. Weed Research 50, 5–24.